A Hartree-Fock-Roothaan Módszer Alkalmazásai a Kvantumkémiában
A kvantumkémia világában kevés olyan módszer létezik, amely annyira alapvetően megváltoztatta volna a molekuláris rendszerek megértését, mint a Hartree-Fock elmélet. Ez a közelítő módszer nemcsak a modern számítási kémia alapkövét jelenti, hanem egyben azt a hidat is, amely összeköti a kvantummechanika elvont matematikai formalizmusát a kémiai valósággal. Minden egyes molekulaorbitál-számítás, minden elektronszerkezet-elemzés és számtalan kémiai jelenség megértése ezen a módszeren alapul vagy abból fejlődött ki. A Hartree-Fock-Roothaan-módszer a kvantummechanikában gyökeredzik. Tisztán és logikusan épül fel a kezdetektől egészen a gyakorlati felhasználásig.
A módszerben semmiféle empíriát nem használunk, és az alapvető fizikai állandókon - a magok és elektronok tömegén, töltésén, valamint a Planck-állandón - kívül más tapasztalati adatunk nincs. Az ilyen számítási módszereket ab initio (a kezdetektől - ezúttal latin) módszereknek nevezik. Az ab initio módszerek, amelyek teljes mértékben elsődleges elveken alapulnak, végül érvényesülnek. James és Coolidge [32] 1933-ban publikáltak egy ab initio számítást, amikor a H₂ molekula kötési energiáját 98%-os pontossággal határozták meg. Ez a számítás egy évet vett igénybe, de sikere növelte a szerzők bizalmát, akik ekkor a szemiemipirikus módszert "a hibák szerencsés kiegyenlítődésének" nevezték, ami azért fordul elő, mert "jelentős fontosságú tényezőket" nem vesz figyelembe [32].
A Hartree-Fock Elmélet Gyökerei és Fejlődése
A húszas évek végén Douglas Hartree brit fizikus olyan problémával szembesült, amely a kvantummechanika egyik legnagyobb kihívását jelentette: hogyan lehet megoldani a többelektronos atomok Schrödinger-egyenletét? Az egyetlen elektront tartalmazó hidrogénatom esetében a megoldás elegáns és pontos, de már a héliumatom két elektronja is olyan bonyolultságot jelent, amely analitikus megoldást nem tesz lehetővé. Hartree zseniális ötlete az volt, hogy minden egyes elektron mozgását egy átlagos potenciálban írja le, amely a többi elektron átlagos eloszlásából származik. Ez a self-consistent field (SCF) megközelítés forradalmi volt, mivel lehetővé tette a többelektronos rendszerek kezelését. Jelentős fejlődés a kvantumkémiában Douglas Hartree által kifejlesztett önkonzisztens mező (SCF) módszerének köszönhetően a kétatomos és számos többatomos molekula pontos hullámfüggvényeinek kiszámításából fakadt [39].
Az elmélet azonban még hiányos volt, mivel nem vette figyelembe az elektronok fermionikus természetét. Vladimir Fock szovjet fizikus 1930-ban egészítette ki Hartree módszerét azzal, hogy bevezette az antiszemmetrikus hullámfüggvények használatát. Ez a módosítás biztosította, hogy a hullámfüggvény megfeleljen a Pauli-elvnek, és így született meg a Hartree-Fock elmélet mai formája. A „szemiemipirikus” kifejezést először Michael Polanyi (1891-1976) és Henry Eyring (1901-1981) [36] [37] használták a elméleti kémiában 1931-ben, kísérletükben, hogy egyesítsék a termodinamikát, a kémiai kinetikát, a kvantummechanikát és a vegyértékelektronok kötéselméletét. Az eyringi és polányi megközelítés, amely a kísérleti eredményekkel való egyesítésre törekedett, hogy potenciális energiafelületeket hozzon létre, megmutatta, hogy lehetséges betekintést nyerni az adiabatikus reakciók mechanizmusába, ami a kémiai reakciók dinamikájával kapcsolatos fontos fogalmakhoz, mint például az átmeneti állapot és az aktivált komplexumhoz vezetett. Már akkor is viták folytak az első elveken alapuló és a szemiemipirikus megközelítések összehasonlításáról.
A Hartree-Fock-Roothaan (HFR) Módszer Alapjai
A Hartree-Fock elmélet lényegében egy olyan matematikai keretrendszer, amely lehetővé teszi, hogy többelektronos atomok és molekulák Schrödinger-egyenletét közelítő módon megoldjuk. Az elmélet központi gondolata, hogy az elektronok közötti bonyolult kölcsönhatásokat egy átlagos térrel helyettesítjük, így az egyébként megoldhatatlan problémát kezelhető formára hozzuk. A hullámfüggvényt a spin-orbitálok termékének antiszimmetrikus lineáris kombinációjaként határozza meg. Az egyelektron-függvényeket "orbitáloknak" nevezik, ami a klasszikus pálya kvantummechanikai analógja [41]. A Hartree-Fock egyenleteket általában iteratív eljárással (SCF) oldják meg. Az eljárás végén megkapjuk a rendszer monoelektronikus HF sajátértékeit. Ezenkívül a teljes elektronenergia (E) is meghatározható az orbitálenergiákból. Ez a teljes energia azonban nem egyszerűen egyenlő az egyelektron-energiák összegével. Ez a tény annak köszönhető, hogy az egyelektron-energiák összege kétszeresen tartalmazza az elektron-elektron kölcsönhatást, azaz az 1. és 2. elektron közötti taszítás mindkét elektronhoz társított egyelektron-energiához hozzájárul.
A Hartree-Fock egyenletek atomokra és molekulákra történő megoldásának problémája a központi szimmetria hiányából adódik. Ezért közelítéseket kell alkalmazni a legjobb orbitál eléréséhez. Így sok elektront tartalmazó rendszerek esetén a Hartree-Fock egyenletek közelítő megoldása K bázisfüggvény lineáris kombinációjára való kiterjesztésből áll, Roothaan [43] javaslatának megfelelően, és ezt a módszert Hartree-Fock-Roothaan (HFR) vagy Molekuláris-Orbitális Lineáris Kombinációs Atomi Orbitáloknak (LCAO-MO) nevezik. Ahol a kiterjesztési együtthatók, amelyeket variációs paraméterként kezelnek, és a bázisfüggvények Slater-típusú atomi orbitálok vagy Gauss-típusúak. Az orbitálok pontos reprezentálásához a függvényeknek teljes halmazt kell alkotniuk. Ez azonban végtelen számú ilyen függvényt igényel. Amit valójában használni kell, az a bázisfüggvények véges száma. Az orbitáloknak meg kell felelniük az ortonormalitási feltételnek. A molekulákra molekuláris orbitálok vonatkoznak, amelyek atomi orbitálok. Így jelentős javulás figyelhető meg a számítási kalkulációkban, amikor az orbitális függvényeket véges bázisfüggvény-készlet szerint terjesztik ki. Az integrál-differenciálegyenletek ezután algebrai egyenletekké alakulnak át az együtthatók kiterjesztéséhez [41]. A legjobb együtthatókat az elektronenergia (26. egyenlet) szerint változtatva határozzák meg, betartva az ortonormalitás feltételét.
Alapvető közelítések és feltételezések
A Hartree-Fock elmélet három alapvető közelítésen nyugszik, amelyek megértése kulcsfontosságú a módszer helyes alkalmazásához:
- Born-Oppenheimer közelítés: Az atommagok mozgása elhanyagolható az elektronokéhoz képest.
- Egypartikulás közelítés: Minden elektron független részecskének tekinthető egy átlagos térben.
- Átlagos tér közelítés: Az elektron-elektron kölcsönhatások átlagolt formában jelennek meg.
- Variációs elv: A hullámfüggvény paramétereit az energia minimalizálásával határozzuk meg.
- Slater-determináns: Az antiszemmetrikus hullámfüggvény matematikai reprezentációja.
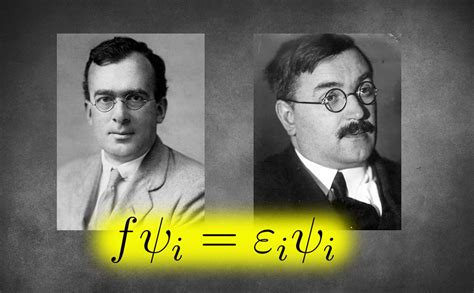
A Fock-operátor a Hartree-Fock elmélet központi eleme, amely tartalmazza az egyelektronos kinetikus energiát, a mag-elektron vonzást, valamint az elektron-elektron taszítás átlagolt hatását. Az egyelektron-integrál az orbitálban lévő elektron kinetikus energiájának és a magok hatása miatt fellépő potenciális integráljának összegét jelenti. A Coulomb-integrálok és az exchange-integrálok egy orbitálban lévő elektron és egy másik orbitálban lévő elektron közötti kölcsönhatáshoz kapcsolódnak. A klasszikus mechanikában a Coulomb-integrál két töltéseloszlás közötti kölcsönhatási energiát jelenti, míg az exchange-integrálnak nincs analógja a klasszikus mechanikában, jelenléte a hullámfüggvény antiszimmetriájából adódik [41].
A bázishalmazok szerepe és típusai
A bázishalmazok kiválasztása kritikus fontosságú a Hartree-Fock számítások pontossága szempontjából. A megfelelő bázishalmaz kiválasztása egyértelműen javítja a számítási teljesítményt. Különböző típusú bázishalmazok eltérő előnyökkel és hátrányokkal rendelkeznek:
| Bázishalmaz típusa | Előnyök | Hátrányok | Alkalmazási terület |
|---|---|---|---|
| Minimális bázis (STO-3G) | Gyors számítás, kis memóriaigény | Alacsony pontosság | Kvalitatív elemzések |
| Split-valence (3-21G) | Jó kompromisszum sebesség és pontosság között | Korlátozott flexibilitás | Rutinszámítások |
| Polarizált bázis (6-31G*) | Jobb kötésleírás | Nagyobb számítási igény | Szerkezeti optimalizálás |
| Diffúz bázis (6-31+G*) | Anionok, gerjesztett állapotok | Lineáris függőség problémák | Speciális rendszerek |
A modern kvantumkémiai számításokban a bázishalmazok fejlesztése folyamatos, és egyre pontosabb leírást tesznek lehetővé a molekuláris tulajdonságok vonatkozásában.
A Hartree-Fock Eljárás Számítógépes Implementációja
A program atomokkal és molekulákkal dolgozik, így elvárható, hogy a programban legyenek olyan objektumok, amelyek ezekkel foglalkoznak. Egy `Atom` osztály tartalmazza az atom helyzetét, rendszámát és elektronszámát. Az atomszám nem feltétlenül egyezik meg az elektronszámmal, mert lehet ion is, de általában a teljes molekula elektronszámát használják. A `Molecule` osztály sok atomot tartalmaz. Ezek az osztályok a `Systems` névtérben találhatók, a csoportosítás és a könnyebb lokalizálás érdekében.
A program atomi orbitálokat használ bázisként. Pontosabban, ezeket egy báziskészlet létrehozására használják, amely nem teljes, de megpróbálja lefedni azt az alteret, amely a valós megoldás nagy részét tartalmazza. Az `Orbital` alaposztály nem kényszerít Gauss-típusú orbitálok (lehetnének Slater-típusúak) vagy kiterjesztett térkoordináták használatára, például gömbi koordinátákra. A program Gauss-orbitálokat használ. Több ilyen orbitált egy „összevont héjba” (contracted shell) csomagolnak, melynek oka az integrálszámítások egyszerűsítése. Egy atom több ilyen héjjal is rendelkezhet. Az `Orbitals` névtérben található `QuantumNumbers` osztály az l, m, n egész értékeket foglalja magában, és generátorként is működik, ami egyszerűsíti az integrálok iterációját a kódban. A `Basis` osztály, amely a `Chemistry` névtérben található, sok atomot csomagol össze a héjaikkal, és textfájlból tölti be azokat. A program jelenleg STO-3G és STO-6G báziskészleteket használ, de minimális módosításokkal más Gauss-báziskészleteket is képes lenne használni.
Mátrixreprezentációt használnak, a program sokat foglalkozik mátrixokkal. Az operátorok mátrixok, és ez a megvalósításban is tükröződik. A `Matrices` névtér tartalmazza az átfedési, kinetikai és nukleáris mátrixokat. Ezek egy `Eigen` mátrixot tartalmaznak, és rendelkeznek `Calculate` metódussal. Az integrálokat az `IntegralRepository`-ból szerzik be. Az implementáció könnyű, de problémát okozhat, hogy melyik vonalak/oszlopok mit képviselnek.
Az integrálok számítása a program legnehezebb része. Az átfedési, kinetikai, nukleáris és elektron-elektron integrálokhoz tartozó osztályok a `GaussianIntegrals` névtérben találhatók. Az elektron-elektron integrál kódjában a rekurziós képletek a publikált irodalommal összhangban vannak. A kód a GitHubon elérhető, és az irodalom elegendő a megértéséhez.
Computational Chemistry 4.14 - Hartree-Fock Approximation
A Hartree-Fock Algoritmus
A Hartree-Fock módszert a `HartreeFock` névtérben implementálták. A megszorított módszer két osztályban van megvalósítva: egy alaposztályban, a `HartreeFockAlgorithm`-ban (amely az unrestricted implementáció alaposztálya is), és a `RestrictedHartreeFock` osztályban. Az önkonzisztens mező iterációja a `Calculate` metódusban van implementálva, amely ciklusban hívja meg a `Step` metódust, amíg az energia konvergál, vagy eléri a maximális iterációk számát. A `Step` implementáció a megszorított módszerre a Fock mátrix inicializálását, a Roothaan-Hall egyenlet megoldását, a sűrűségmátrix kiszámítását és az energia frissítését tartalmazza. A `NormalizeC` hívás nem feltétlenül szükséges, de gyorsabb konvergenciát eredményezhet, és fizikailag is értelmes a sűrűségmátrix normalizálása. A `InitFockMatrix` implementálja a Fock mátrix inicializálását.
A Korrelációs Energia Problémája és Korlátai
A Hartree-Fock elmélet egyik legnagyobb korlátja, hogy nem veszi figyelembe az elektron-elektron korrelációt. Ez alatt azt értjük, hogy a valóságban az elektronok mozgása korrelált: ha az egyik elektron egy adott helyen tartózkodik, az befolyásolja a többi elektron valószínű tartózkodási helyét. A korrelációs energia definíció szerint a pontos elektronenergia és a Hartree-Fock energia közötti különbség: E_corr = E_exact - E_HF. Ez az energia általában a teljes kötési energia 1-3%-át teszi ki, de ez a látszólag kis érték gyakran nagyobb, mint maga a kötési energia, így elhanyagolása súlyos hibákhoz vezethet.
Az elektronkorreláció két fő típusra osztható: dinamikus és statikus korrelációra. A dinamikus korreláció az elektronok pillanatnyi taszításából származik, míg a statikus korreláció a degenerált vagy közel degenerált orbitálok keverésével kapcsolatos. A dinamikus korreláció hatásai különösen fontosak a kötési energiák pontos számítása, a molekulageometriák optimalizálása, a rezgési frekvenciák meghatározása és a termokémiai adatok számítása esetében.
Post-Hartree-Fock Módszerek Áttekintése
A Hartree-Fock elmélet korlátainak felismerése vezetett a post-Hartree-Fock módszerek fejlesztéséhez, amelyek célja a korrelációs energia figyelembevétele. Ezek a módszerek a Hartree-Fock hullámfüggvényt kiindulópontként használják, és különböző matematikai technikákkal finomítják azt.
Konfigurációs kölcsönhatás (CI) módszerek
A Configuration Interaction (CI) módszerek a hullámfüggvényt különböző elektronkonfigurációk lineáris kombinációjaként írják fel. A teljes CI (Full CI) elméletileg pontos megoldást ad, de gyakorlatilag csak nagyon kis rendszerekre alkalmazható. A gyakorlatban gyakran használt közelítések a CIS (egyszeresen gerjesztett konfigurációk), CISD (egyszeresen és kétszeresen gerjesztett konfigurációk) és CISDT (háromszorosan gerjesztett konfigurációkat is tartalmaz) módszerek.
Perturbációelmélet alapú módszerek
A Møller-Plesset perturbációelmélet (MP) egy másik megközelítést képvisel a korreláció kezelésére. A módszer a Hartree-Fock energiát nulladrendű közelítésnek tekinti, és perturbációs sorfejtéssel számítja ki a korrekciót. A leggyakrabban használt szintek az MP2 (másodrendű perturbációs korrekció), MP3 (harmadrendű korrekció) és MP4 (negyedrendű korrekció).
Sűrűségfunkcionál-elmélet (DFT) Kapcsolata
Bár a Density Functional Theory (DFT) formálisan különbözik a Hartree-Fock elmélettől, gyakorlati megvalósításában sok közös elemet tartalmaz. A DFT alapgondolata, hogy a rendszer összes tulajdonsága kifejezhető az elektronsűrűség függvényeként.
A Kohn-Sham egyenletek és hasonlóságok
A DFT gyakorlati alkalmazása a Kohn-Sham formalizmus keretében történik, amely matematikailag nagyon hasonlít a Hartree-Fock egyenletekhez. A fő különbség az exchange-korrelációs funkcionál használata, amely implicit módon tartalmazza a korrelációs hatásokat. A Kohn-Sham egyenletek a következőképpen írhatók fel: [ĥ + v_H + v_XC]φᵢ = εᵢφᵢ, ahol v_H a Hartree-potenciál, v_XC pedig az exchange-korrelációs potenciál.
Hibrid funkcionálok és a Hartree-Fock exchange
A modern DFT számításokban gyakran használnak hibrid funkcionálokat, amelyek a Hartree-Fock exchange egy részét kombinálják a DFT exchange-korrelációs funkcionálokkal. A legnépszerűbb hibrid funkcionál a B3LYP, amely 20% Hartree-Fock exchange-et tartalmaz.

Alkalmazási Területek és Példák
Kémiai szimulációk végrehajtásához a szükséges feltétel az adott fizikai jelenség ismerete, a megfelelő számítástechnikai módszerek választéka, egy nagy teljesítményű számítógép - valamint a megfelelő szakértelem. A kvantumkémia központi fontosságú a Schrödinger-egyenlet megoldásainak megszerzésében az atomi és molekuláris rendszerek tulajdonságainak pontos meghatározásához. A fizikai kémia területén belül a kvantumkémia különféle termodinamikai tulajdonságok, például az entrópia, a gázok hőkapacitásának kiszámítására, a molekuláris spektrumok értelmezésére, a kötéshosszak, kötésszögek, dipólusmomentum számítására és az intermolekuláris erők megértésére szolgál.
Az organikus kvantumkémiában becsülheti a molekulák stabilitását, kiszámíthatja a közbenső reakciók tulajdonságait, reprodukálhatja a szerves vegyületek aromatikusságát, és szimulálhatja a nukleáris mágneses rezonancia (NMR) spektrumokat. Az analitikai kémiában a spektrális vonalak intenzitásának értelmezésére használják. A szervetlen kémia területén a ligandumtér-elmélethez használják, ahol előrejelzéseket tesznek, és tulajdonságokat igazolnak az átmeneti fémek komplex ionjaira.
Szerkezeti kémia és molekulageometria
A Hartree-Fock elmélet egyik legsikeresebb alkalmazási területe a molekulageometriák meghatározása. A módszer általában jó pontossággal reprodukálja a kötéshosszakat és kötésszögeket, bár a kötési energiáknál kevésbé megbízható. Tipikus alkalmazások az egyensúlyi geometriák optimalizálása, konformációs analízis, átmeneti állapotok keresése és reakcióutak feltérképezése.
Bioaktív vegyületek tervezése
A kvantumkémiai módszerek alkalmazása a bioaktív vegyületek tanulmányozásában és tervezésében ma már elterjedt gyakorlattá vált. Pinheiro, Ferreira és Romero (2001) kombinált kvantumkémiai (Hartree-Fock 3-21G) és multivariáns analízis módszereket (PCA, HCA, KNN és SIMCA) alkalmaztak dihidroartemizin származékok tanulmányozására és javaslatára. Artemisinin-származékokat antimaláriás aktivitással a Plasmodium falciparum ellen, amely rezisztens a mefloquine-ra, kvantumkémiai módszerek (HF/6-31G*) és a parciális legkisebb négyzetek (PLS) módszerével vizsgáltak. Egy 10 javasolt artemisinin-származékból (ismeretlen antimaláriás aktivitással a Plasmodium falciparum ellen) egy új vegyületet állítottak elő, amely a korábban irodalomban leírt vegyületekhez képest kiváló antimaláriás aktivitással rendelkezett [20]. Cardoso et al. (2008) artemisinint és néhány származékát vizsgálták, amelyek aktivitást mutatnak a Plasmodium falciparum D-6 törzsei ellen a HF/3-21G módszerrel. Az MEP-et arra használták, hogy azonosítsák azokat a kulcsfontosságú tulajdonságokat, amelyek szükségesek a vegyületek aktivitásához, és ezeket felhasználva új artemisinin-származékokat javasoltak [21].
Spektroszkópiai tulajdonságok számítása
A Hartree-Fock orbitálok kiváló kiindulópontot jelentenek különböző spektroszkópiai tulajdonságok számításához, mint például az NMR kémiai eltolódások (a mágneses árnyékolási tenzor számítása), IR spektrumok (rezgési frekvenciák és intenzitások), UV-Vis spektrumok (gerjesztési energiák post-HF módszerekkel) és fotoelektron spektrumok (ionizációs potenciálok a Koopmans-tétel alapján).
Oktatási Jelentőség és Gyakorlati Tanácsok
A Hartree-Fock elmélet pedagógiai érteke felbecsülhetetlen a kvantumkémia tanításában. A módszer átlátható szerkezete lehetővé teszi a kvantummechanikai alapfogalmak megértését anélkül, hogy elveszne a matematikai részletekben. A tanulók számára fontos koncepciók közé tartozik a molekulaorbitál-elmélet, a variációs elv alkalmazása, az SCF iteráció megértése és az elektronkorreláció jelentősége. A Hartree-Fock számítások benchmark szerepet töltenek be: minden új módszert ehhez viszonyítanak, és az új technikák fejlesztésénél mindig figyelembe veszik, hogy mennyivel jobbak a HF eredményeknél.
Mikor használjuk a Hartree-Fock módszert?
A Hartree-Fock elmélet ma is hasznos lehet bizonyos esetekben:
- Alkalmas alkalmazások: Nagy rendszerek előzetes vizsgálata, kvalitatív molekulaorbitál-analízis, geometria-optimalizálás kiindulópontja, oktatási célú demonstrációk, módszerfejlesztés tesztelése.
- Kerülendő esetek: Pontos kötési energiák számítása, gyenge kölcsönhatások vizsgálata, nyitott héjú radikálok, erős korrelációjú rendszerek.
tags: #hartley #fock #modell #alkalmazas




