A Hartree-Fock Modell Alkalmazása a Kvantumkémiában: Alapoktól az Alkalmazásokig
A kvantumkémia világában kevés olyan módszer létezik, amely annyira alapvetően megváltoztatta volna a molekuláris rendszerek megértését, mint a Hartree-Fock elmélet. Ez a közelítő módszer nemcsak a modern számítási kémia alapkövét jelenti, hanem egyben azt a hidat is, amely összeköti a kvantummechanika elvont matematikai formalizmusát a kémiai valósággal. Minden egyes molekulaorbitál-számítás, minden elektronszerkezet-elemzés és számtalan kémiai jelenség megértése ezen a módszeren alapul vagy abból fejlődött ki. A Hartree-Fock elmélet lényegében egy olyan matematikai keretrendszer, amely lehetővé teszi, hogy többelektronos atomok és molekulák Schrödinger-egyenletét közelítő módon megoldjuk. Az elmélet központi gondolata, hogy az elektronok közötti bonyolult kölcsönhatásokat egy átlagos térrel helyettesítjük, így az egyébként megoldhatatlan problémát kezelhető formára hozzuk. Ez a megközelítés számos nézőpontból vizsgálható: a matematikai elegancia, a fizikai megalapozottság és a gyakorlati alkalmazhatóság szempontjából egyaránt. Ebben az átfogó elemzésben betekintést nyerhetsz a Hartree-Fock elmélet mélyebb rétegeibe, megismerheted a módszer matematikai hátterét, gyakorlati alkalmazásait és korlátait. Megtudhatod, hogyan kapcsolódik ez az elmélet a modern kvantumkémiai módszerekhez, és milyen szerepet játszik a mai napig a molekuláris rendszerek számítógépes modellezésében.
A Hartree-Fock elmélet történeti háttere és fejlődése
A húszas évek végén Douglas Hartree brit fizikus olyan problémával szembesült, amely a kvantummechanika egyik legnagyobb kihívását jelentette: hogyan lehet megoldani a többelektronos atomok Schrödinger-egyenletét? Az egyetlen elektront tartalmazó hidrogénatom esetében a megoldás elegáns és pontos, de már a héliumatom két elektronja is olyan bonyolultságot jelent, amely analitikus megoldást nem tesz lehetővé. Hartree zseniális ötlete az volt, hogy minden egyes elektron mozgását egy átlagos potenciálban írja le, amely a többi elektron átlagos eloszlásából származik. Ez a self-consistent field (SCF) megközelítés forradalmi volt, mivel lehetővé tette a többelektronos rendszerek kezelését. Az elmélet azonban még hiányos volt, mivel nem vette figyelembe az elektronok fermionikus természetét. Vladimir Fock szovjet fizikus 1930-ban egészítette ki Hartree módszerét azzal, hogy bevezette az antiszemmetrikus hullámfüggvények használatát. Ez a módosítás biztosította, hogy a hullámfüggvény megfeleljen a Pauli-elvnek, és így született meg a Hartree-Fock elmélet mai formája.
Az elmélet matematikai alapjai és központi fogalmai
Az alapvető közelítések és feltételezések
A Hartree-Fock elmélet három alapvető közelítésen nyugszik, amelyek megértése kulcsfontosságú a módszer helyes alkalmazásához:
- 🔬 Born-Oppenheimer közelítés: Az atommagok mozgása elhanyagolható az elektronokéhoz képest
- ⚛️ Egypartikulás közelítés: Minden elektron független részecskének tekinthető egy átlagos térben
- 🧮 Átlagos tér közelítés: Az elektron-elektron kölcsönhatások átlagolt formában jelennek meg
- 📊 Variációs elv: A hullámfüggvény paramétereit az energia minimalizálásával határozzuk meg
- 🎯 Slater-determináns: Az antiszemmetrikus hullámfüggvény matematikai reprezentációja
A Hartree-Fock egyenletek megoldása iteratív folyamat, amely során az orbitálok önkonzisztens módon kerülnek meghatározásra. Ez azt jelenti, hogy minden egyes elektron orbitálja függ a többi elektron orbitáljaitól, és a megoldást addig finomítjuk, amíg ez a függőség konzisztenssé nem válik.
A Fock-operátor és a kanonikus orbitálok
A Fock-operátor a Hartree-Fock elmélet központi eleme, amely tartalmazza az egyelektronos kinetikus energiát, a mag-elektron vonzást, valamint az elektron-elektron taszítás átlagolt hatását. Matematikailag ez az operátor a következő formában írható fel:
F̂ = ĥ + Σⱼ(Ĵⱼ - K̂ⱼ)
ahol ĥ az egyelektronos Hamilton-operátor, Ĵⱼ a Coulomb-operátor és K̂ⱼ az exchange-operátor. Ez a közelítés praktikus megoldást nyújt a molekuláris rendszerek kezelésére, és lehetővé teszi a kémiai kötések kvantummechanikai leírását. Az LCAO módszerben minden molekulaorbitál φᵢ felírható a következő formában:
φᵢ = Σμ cμᵢ χμ
ahol χμ az atomi bázisfüggvények, cμᵢ pedig a megfelelő együtthatók. Ezek az együtthatók határozzák meg, hogy az egyes atomi orbitálok milyen mértékben járulnak hozzá a molekulaorbitálhoz.

A bázishalmazok szerepe és típusai
A bázishalmazok kiválasztása kritikus fontosságú a Hartree-Fock számítások pontossága szempontjából. A különböző típusú bázishalmazok eltérő előnyökkel és hátrányokkal rendelkeznek:
| Bázishalmaz típusa | Előnyök | Hátrányok | Alkalmazási terület |
|---|---|---|---|
| Minimális bázis (STO-3G) | Gyors számítás, kis memóriaigény | Alacsony pontosság | Kvalitatív elemzések |
| Split-valence (3-21G) | Jó kompromisszum sebesség és pontosság között | Korlátozott flexibilitás | Rutinszámítások |
| Polarizált bázis (6-31G*) | Jobb kötésleírás | Nagyobb számítási igény | Szerkezeti optimalizálás |
| Diffúz bázis (6-31+G*) | Anionok, gerjesztett állapotok | Lineáris függőség problémák | Speciális rendszerek |
A modern kvantumkémiai számításokban a bázishalmazok fejlesztése folyamatos, és egyre pontosabb leírást tesznek lehetővé a molekuláris tulajdonságok vonatkozásában.
Gyakorlati alkalmazás: A vízmolekula Hartree-Fock számítása
Vegyük példaként a vízmolekula (H₂O) Hartree-Fock számítását, amely jól szemlélteti a módszer gyakorlati alkalmazását. Ez a számítás lépésről lépésre bemutatja, hogyan jutunk el a molekula elektronszerkezetének leírásához.
Első lépés: Geometria megadása és bázishalmaz választása
Kezdjük a vízmolekula geometriájának megadásával. A kísérleti adatok alapján az O-H kötéshossz 0.96 Å, a H-O-H kötésszög pedig 104.5°. Koordináta-rendszerünkben az oxigénatomot helyezzük az origóba, és a molekulát az xz-síkba orientáljuk. Bázishalmazként válasszuk a 6-31G* halmazt, amely megfelelő pontosságot biztosít a vízmolekula leírásához. Ez a bázishalmaz 13 bázisfüggvényt tartalmaz: az oxigénatomon 9-et (1s, 2s, 2px, 2py, 2pz, és d-polarizációs függvények), minden hidrogénatomra pedig 2-2 függvényt (1s és p-polarizációs).
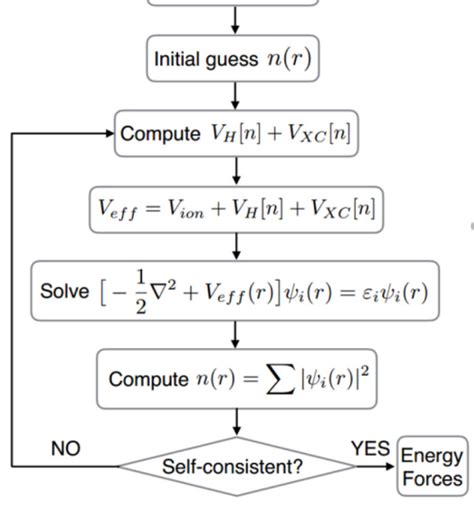
Második lépés: Kezdeti becslés és SCF iteráció
A számítás kezdetekor szükségünk van egy kezdeti becslésre a molekulaorbitálokra vonatkozóan. Ezt általában a magányos atomok orbitáljainak szuperpozíciójával (SOAD - Sum of Atomic Orbital Densities) kapjuk meg. Az SCF iteráció során a következő lépéseket ismételjük:
- Sűrűségmátrix kiszámítása a jelenlegi orbitálokból
- Fock-mátrix felépítése a kétcentrumos integrálok felhasználásával
- Fock-egyenlet megoldása új orbitálok meghatározásához
- Konvergencia ellenőrzése az energia és sűrűség változása alapján
Harmadik lépés: Eredmények értelmezése
A konvergált számítás eredményeként megkapjuk a vízmolekula molekulaorbitáljait és azok energiáit. A vízmolekulában 10 elektron található, amelyek 5 elfoglalt molekulaorbitált töltenek fel:
| Orbitál | Energia (hartree) | Jelleg | Leírás |
|---|---|---|---|
| 1a₁ | -20.55 | O 1s | Oxigén belső héj |
| 2a₁ | -1.35 | O 2s | Oxigén vegyérték s |
| 1b₂ | -0.69 | O 2py | Oxigén p orbital |
| 3a₁ | -0.57 | Kötő | O-H kötések |
| 1b₁ | -0.50 | Magányos pár | Oxigén magányos elektronpár |

Korlátok és kihívások
Gyakori hibák és elkerülésük
A Hartree-Fock számítások során több tipikus hiba fordulhat elő, amelyek felismerése és elkerülése fontos a megbízható eredmények eléréséhez:
- Konvergencia problémák: Túl nagy kezdeti lépésméret vagy rossz kezdeti becslés miatt
- Bázishalmaz hiányosságok: Túl kicsi bázishalmaz nem megfelelő leírást ad
- Szimmetria megszakadás: A molekula szimmetriájának figyelmen kívül hagyása
- Spin-szennyezés: Nyitott héjú rendszerekben a különböző spin-állapotok keveredése
„A Hartree-Fock elmélet nem a tökéletes megoldás, hanem egy kiváló kiindulópont a molekuláris rendszerek megértéséhez.”
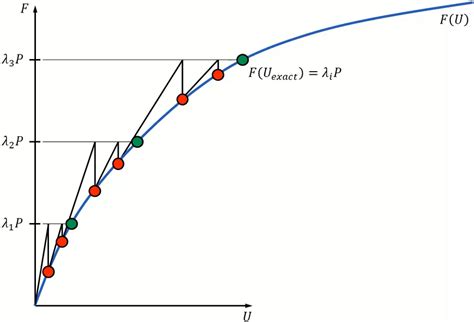
A korrelációs energia problémája és korlátai
A Hartree-Fock elmélet egyik legnagyobb korlátja, hogy nem veszi figyelembe az elektron-elektron korrelációt. Ez alatt azt értjük, hogy a valóságban az elektronok mozgása korrelált: ha az egyik elektron egy adott helyen tartózkodik, az befolyásolja a többi elektron valószínű tartózkodási helyét. A korrelációs energia definíció szerint a pontos elektronenergia és a Hartree-Fock energia közötti különbség:
Ecorr = Eexact - EHF
Ez az energia általában a teljes kötési energia 1-3%-át teszi ki, de ez a látszólag kis érték gyakran nagyobb, mint maga a kötési energia, így elhanyagolása súlyos hibákhoz vezethet. Az elektronkorreláció két fő típusra osztható: dinamikus és statikus korrelációra. A dinamikus korreláció az elektronok pillanatnyi taszításából származik, míg a statikus korreláció a degenerált vagy közel degenerált orbitálok keverésével kapcsolatos. A dinamikus korreláció hatásai különösen fontosak a következő esetekben:
- Kötési energiák pontos számítása
- Molekulageometriák optimalizálása
- Rezgési frekvenciák meghatározása
- Termokémiai adatok számítása
„A korrelációs energia figyelmen kívül hagyása olyan, mintha egy órát másodpercek nélkül próbálnánk használni - működik, de nem elég pontos a precíz munkához.”
Túl a Hartree-Fockon: Korrelált módszerek és DFT
Post-Hartree-Fock módszerek áttekintése
A Hartree-Fock elmélet korlátainak felismerése vezetett a post-Hartree-Fock módszerek fejlesztéséhez, amelyek célja a korrelációs energia figyelembevétele. Ezek a módszerek a Hartree-Fock hullámfüggvényt kiindulópontként használják, és különböző matematikai technikákkal finomítják azt.
Konfigurációs kölcsönhatás (CI) módszerek
A Configuration Interaction (CI) módszerek a hullámfüggvényt különböző elektronkonfigurációk lineáris kombinációjaként írják fel. A teljes CI (Full CI) elméletileg pontos megoldást ad, de gyakorlatilag csak nagyon kis rendszerekre alkalmazható. A gyakorlatban gyakran használt közelítések:
- CIS: Egyszeresen gerjesztett konfigurációk
- CISD: Egyszeresen és kétszeresen gerjesztett konfigurációk
- CISDT: Háromszorosan gerjesztett konfigurációkat is tartalmaz
Perturbációelmélet alapú módszerek
A Møller-Plesset perturbációelmélet (MP) egy másik megközelítést képvisel a korreláció kezelésére. A módszer a Hartree-Fock energiát nulladrendű közelítésnek tekinti, és perturbációs sorfejtéssel számítja ki a korrekciót. A leggyakrabban használt szintek:
- MP2: Másodrendű perturbációs korrekció
- MP3: Harmadrendű korrekció
- MP4: Negyedrendű korrekció
„A post-Hartree-Fock módszerek olyan, mint a finom hangolás egy zenei hangszeren - a Hartree-Fock adja az alapot, de a részletek teszik tökéletessé a harmóniát.”
Sűrűségfunkcionál-elmélet (DFT) kapcsolata
Bár a Density Functional Theory (DFT) formálisan különbözik a Hartree-Fock elmélettől, gyakorlati megvalósításában sok közös elemet tartalmaz. A DFT alapgondolata, hogy a rendszer összes tulajdonsága kifejezhető az elektronsűrűség függvényeként.
A Kohn-Sham egyenletek és hasonlóságok
A DFT gyakorlati alkalmazása a Kohn-Sham formalizmus keretében történik, amely matematikailag nagyon hasonlít a Hartree-Fock egyenletekhez. A fő különbség az exchange-korrelációs funkcionál használata, amely implicit módon tartalmazza a korrelációs hatásokat. A Kohn-Sham egyenletek:
[ĥ + v_H + v_XC]φᵢ = εᵢφᵢ
ahol v_H a Hartree-potenciál, v_XC pedig az exchange-korrelációs potenciál.
Hibrid funkcionálok és a Hartree-Fock exchange
A modern DFT számításokban gyakran használnak hibrid funkcionálokat, amelyek a Hartree-Fock exchange egy részét kombinálják a DFT exchange-korrelációs funkcionálokkal. A legnépszerűbb hibrid funkcionál a B3LYP, amely 20% Hartree-Fock exchange-et tartalmaz. „A hibrid funkcionálok azt mutatják, hogy a Hartree-Fock elmélet még a modern kvantumkémiában is nélkülözhetetlen építőelem.”
Számítási implementáció és optimalizálás
Algoritmusok és számítási komplexitás
A Hartree-Fock számítások implementálása során a legkritikusabb pont a kétcentrumos integrálok kiszámítása és tárolása. Ezek száma N⁴-nel skálázódik a bázisfüggvények számával, ami nagy rendszerek esetén jelentős számítási kihívást jelent. Modern implementációkban különböző optimalizálási technikákat alkalmaznak:
- 🔧 Integrál screening: Kis értékű integrálok elhanyagolása
- ⚡ Direct SCF: Integrálok újraszámítása tárolás helyett
- 🖥️ Párhuzamosítás: Többprocesszoros architektúrák kihasználása
- 💾 Memória menedzsment: Hatékony adatstruktúrák használata
- 🎯 Konvergencia gyorsítás: DIIS és hasonló technikák alkalmazása

A DIIS konvergencia gyorsítás
A Direct Inversion in the Iterative Subspace (DIIS) módszer jelentősen felgyorsítja az SCF konvergenciát azáltal, hogy a korábbi iterációk információit felhasználja a következő becslés javítására. Ez különösen fontos nehezen konvergáló rendszerek esetében.
Alkalmazási területek és példák
Szerkezeti kémia és molekulageometria
A Hartree-Fock elmélet egyik legsikeresebb alkalmazási területe a molekulageometriák meghatározása. A módszer általában jó pontossággal reprodukálja a kötéshosszakat és kötésszögeket, bár a kötési energiáknál kevésbé megbízható. Tipikus alkalmazások:
- Egyensúlyi geometriák optimalizálása
- Konformációs analízis
- Átmeneti állapotok keresése
- Reakcióutak feltérképezése

Spektroszkópiai tulajdonságok számítása
A Hartree-Fock orbitálok kiváló kiindulópontot jelentenek különböző spektroszkópiai tulajdonságok számításához:
- NMR kémiai eltolódások: A mágneses árnyékolási tenzor számítása
- IR spektrumok: Rezgési frekvenciák és intenzitások
- UV-Vis spektrumok: Gerjesztési energiák (post-HF módszerekkel)
- Fotoelektron spektrumok: Ionizációs potenciálok (Koopmans-tétel)
„A Hartree-Fock elmélet olyan, mint egy jó térkép - nem minden részletet mutat meg, de megbízhatóan eligazít a kvantumkémiai tájban.”
Termokémiai számítások
Bár a Hartree-Fock energia önmagában nem alkalmas pontos termokémiai adatok számítására, izodesmic reakciók esetében a hibák gyakran kioltják egymást, és így meglepően jó eredményeket kaphatunk. Napjainkban a Hartree-Fock elmélet ritkán használatos önálló módszerként, inkább más fejlettebb technikák alapjaként szolgál. Minden modern kvantumkémiai programcsomag tartalmazza, és a legtöbb számítás ezzel a módszerrel kezdődik.
Gyógyszerkutatási alkalmazások
Az ab initio módszerek, mint a Hartree-Fock, alkalmazása a bioaktív vegyületek tervezésében és vizsgálatában mindennapos gyakorlattá vált. Pinheiro, Ferreira és Romero (2001) például kvantumkémiai (Hartree-Fock 3-21G) és multivariáns analízis módszereket kombinálva tanulmányozták és javasoltak dihidroartemisinin származékokat. Későbbi kutatásokban artemisinin származékokat, amelyek antimaláriás aktivitással rendelkeznek a meflokin-rezisztens Plasmodium falciparum ellen, kvantumkémiai módszerekkel (HF/6-31G*) és részleges legkisebb négyzetek (PLS) módszerrel vizsgáltak. Egy 10 javasolt artemisinin származékból álló készletből egy új vegyületet állítottak elő, amely kiváló antimaláriás aktivitást mutatott a korábban leírt vegyületekhez képest. Cardoso és társai (2008) artemisinint és egyes származékait tanulmányozták a Plasmodium falciparum D-6 törzsei elleni aktivitásuk szempontjából, a HF/3-21G módszert alkalmazva. A kapott geometria megbízhatóságának ellenőrzésére összehasonlították az artemisinin trioxán gyűrűjének szerkezeti paramétereit az irodalomból származó elméleti és kísérleti értékekkel. Molekuláris elektrosztatikus potenciált (MEP) használtak a vegyületek aktivitásához szükséges kulcsfontosságú jellemzők azonosítására, és ezeket felhasználták új artemisinin származékok javaslatához.
Az ab initio megközelítés jelentősége
A Hartree-Fock-Roothaan-módszer a kvantummechanikában gyökerezik. Tisztán és logikusan épül fel a kezdetektől egészen a gyakorlati felhasználásig. A módszerben semmiféle empíriát nem használunk, és az alapvető fizikai állandókon - a magok és elektronok tömegén, töltésén, valamint a Planck-állandón - kívül más tapasztalati adatunk nincs. Az ilyen számítási módszereket ab initio (a kezdetektől - ezúttal latin) módszereknek nevezik. Ab initio HFR-számításokhoz tehát mindössze az atomok kiindulási elrendezésére és egy bázis kiválasztására van szükség, no és persze egy működő számítógépes programra. Megfelelő programok ma már rendelkezésre állnak, használatuk néhány óra alatt elsajátítható. Akkor miért ne számoljunk?
Oktatási jelentőség és benchmark szerep
Oktatási jelentőség
A Hartree-Fock elmélet pedagógiai érteke felbecsülhetetlen a kvantumkémia tanításában. A módszer átlátható szerkezete lehetővé teszi a kvantummechanikai alapfogalmak megértését anélkül, hogy elveszne a matematikai részletekben. A tanulók számára fontos koncepciók:
- Molekulaorbitál-elmélet
- Variációs elv alkalmazása
- SCF iteráció megértése
- Elektronkorreláció jelentősége
Benchmark szerepe
A Hartree-Fock számítások benchmark szerepet töltenek be: minden új módszert ehhez viszonyítanak, és az új technikák fejlesztésénél mindig figyelembe veszik, hogy mennyivel jobbak a HF eredményeknél. „A Hartree-Fock elmélet nem azért fontos, mert tökéletes, hanem azért, mert megérthetővé teszi a kvantumkémia alapjait.”
Gyakorlati tanácsok és ajánlások
Mikor használjuk a Hartree-Fock módszert?
A Hartree-Fock elmélet ma is hasznos lehet bizonyos esetekben:
Alkalmas alkalmazások:
- Nagy rendszerek előzetes vizsgálata
- Kvalitatív molekulaorbitál-analízis
- Geometria-optimalizálás kiindulópontja
- Oktatási célú demonstrációk
- Módszerfejlesztés tesztelése
Kerülendő esetek:
- Pontos kötési energiák számítása
- Gyenge kölcsönhatások vizsgálata
- Nyitott héjú radikálok
- Erős korrelációjú rendszerek
Bázishalmaz választási útmutató
A megfelelő bázishalmaz kiválasztása kulcsfontosságú a számítás pontossága és hatékonysága szempontjából. Mindig figyelembe kell venni a vizsgált rendszer méretét és a kívánt pontosságot. Kisebb rendszerek és kvalitatív eredmények esetén minimális bázishalmazok is elegendőek lehetnek, míg pontosabb vizsgálatokhoz polarizált vagy diffúz bázishalmazok alkalmazása javasolt.
tags: #hartree #fock #modell #alkalmazas




